
بقلم الدكتور المهندس محمد ملحم*
تمهيد
تشير مواقع العمل إلى أنّ الدخل السنوي سيبلغ حدود 80 ألف دولار لمن سيحمل لقب أخصائي معلوماتية حيوية، وتتوقع أحدث التقارير أيضاً أن تنمو الوظائف المرتبطة بالمعلوماتية الحيوية بنسبة 15٪ بحلول عام 2029. لقد أصبحت المعلوماتية الحيوية (كفرع من فروع التحليل القائم على الحاسب) بمتناول الأفراد بعد أن كانت حبيسة المختبرات البحثية، وإن القدرة على امتلاك أجهزة حاسب متطورة ساعد على تسريع البحث في هذا المجال وتقديم اكتشافات جديدة. تقدم هذه المقالة نظرة عامة عن المعلوماتية الحيوية، تشمل قواعد البيانات البيولوجية ومشاكلها، وطرق وخوارزميات تحليل البيانات الحيوية، والآفاق المستقبلية للمعلوماتية الحيوية في عدة مجالات.
1-مقدمة
المعلوماتية الحيوية (Bioinformatics): هي مجال بحث متعدّد التّخصصات، يجمع بين علوم الحاسب والرياضيات والعلوم البيولوجية والإحصاء. فقد عرّف المعهد العالمي للصحة (NIH) المعلوماتية الحيوية، على أنها اتحاد للبيولوجيا والمعلوماتية، وتنطوي المعلوماتية الحيوية على التكنولوجيا التي تستخدم الحواسب للتخزين والاسترجاع والمعالجة وتوزيع المعلومات المتعلقة بالجزيئات البيولوجية الحيوية مثل (DNA) و(RNA) والبروتينات. أما المركز الوطني لمعلومات التكنولوجيا الحيوية (NCBI)، فقد حدّد المعلوماتية الحيوية في ثلاثة مسارات [1,2]:
- طرح خوارزميات وطرق احصاء جديدة لتخمين العلاقة بين الكم الهائل من البيانات.
- تحليل وتفسير مختلف أنواع البيانات (مثل النيكلوتيدات والأحماض الأمينية، والبروتينات وبنيتها).
- تطوير أدوات قادرة على إدارة واستكشاف مختلف صنوف المعلومات.
تهدف المعلوماتية الحيوية إلى فهم آلية عمل الخليّة الحيّة على المستوى الجزيئي، أي كيف يتم نسخ الحمض النووي DNA إلى الحمض النووي الريبي RNA ومن ثم ترجمته إلى البروتينات، فتركز عبر أدواتها على تحليل التتابع الجزيئي، والتحليل الهيكلي الجزيئي، والتحليل الوظيفي الجزيئي. ومن أهم تطبيقاتها، دراسة تتابعات الحمض النووي DNA، وتحليل الجين، والبيولوجيا التطورية evolutionary biology، والمعلوماتية المناعية immunoinformatics والتعبير الجيني gene expression ودراسة البروتينات وطرق إنتاجها في الكائن الحي proteomics. وهناك مهام أخرى تتعلق بالنواحي الفيزيائية مثل التنبّؤ بالطاقة اللازمة لعمليات التداخل الفيزيائي للجزيئات وإمكانية ارتباطها وتستخدم في تصميم الأدوية. وهناك تطبيقات أخرى في التكنولوجيا الحيوية الزراعية والطب الشرعي [3].
2-قواعد البيانات البيولوجية
يقول ستيفان كالو زياك، الأستاذ المساعد في المعلوماتية الحيوية في جامعة نورث إيسترن: “لدينا بيانات أكثر مما نعرف ماذا نفعل بها” هذه البيانات موزعة في قواعد بيانات بيولوجية. تستخدم هذه القواعد جميع أنواع الهياكل المعروفة مثل قواعد البيانات العلائقية والشيئيّة الموجهة والهرميّة والشبكيّة، وتتوزع هذه القواعد في العديد من مراكز المعلوماتية الحيوية حول العالم، ولعلّ أهمها، مركز NCBI الأمريكي ومعهد EBI2 البريطاني ومخبر EMBL الألماني ومركز DDBJ4 الياباني. تتوزع قواعد البيانات البيولوجية حول العالم وفق الشكل (1) [2, 4].
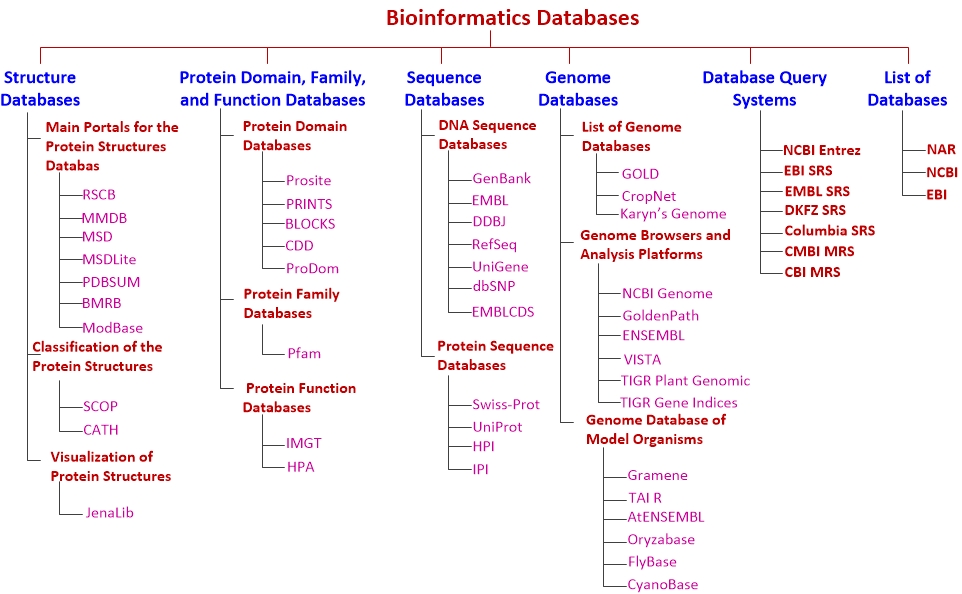
وتُصنّف في ثلاث فئات:
1-قواعد بيانات أوليّة
تحتوي على بيانات بيولوجية أصلية، وأهم هذه القواعد GenBank أو EMBL أو DDBJ، تخزّن بيانات تتابع الحمض النووي الخام التي ينتجها الباحثون في جميع أنحاء العالم، وكلها متاحة مجاناً على شبكة الإنترنت. ويُعد حالياً، تقديم التتابع الجيني إلى أحد هذه القواعد شرطاً مسبقاً للنشر في المجلات العلمية.
2-قواعد بيانات ثانوية
وتحتوي على معلومات معالجة حاسوبياً، أو منسّقة يدوياً استناداً إلى المعلومات الأصلية من قواعد البيانات الأولية. تختلف قواعد البيانات الثانوية فيما بينها بشكل كبير، فالبعض منها يكون بمثابة أرشيف بسيط من تتابعات مترجمة عن DNA. والبعض الآخر يقدّم مستوياتٍ أعلى من المعلومات عن التركيب والوظيفة. من الأمثلة البارزة على قواعد البيانات الثانوية Swiss-Port،
و TrEMBL، و PIR.
3-قواعد بيانات متخصّصة
وتخدم مجتمعاً بحثياً مُعيّناً أو تركّز على كائن معين. وقد يكون محتوى هذه القواعد تتابعات أو أنواع أخرى من المعلومات. قد تتداخل مع قاعدة بيانات أساسية، أو قد تحتوي على بيانات جديدة مقدّمة مباشرة من قبل المؤلفين. وتتضمّن أمثلة مثل Fly base، و WormBas، وTAIR.
مشكلات قواعد البيانات البيولوجية:
تتلخص المشاكل المرتبطة بقواعد البيانات البيولوجية بالآتي:
–الاعتماد المفرط على المعلومات الخاصة بالتتابعات والشروحات غير الموثّقة المرافقة لها.
-تجاهل حقيقة وجود العديد من الأخطاء في قواعد البيانات أثناء تحديد التتابعات.
-المستويات العالية من التكرار في قواعد بيانات التتابعات الأساسي، يبين الشكل (2) الانخفاض الحاد في إدخالات TrEMBL مع إجراء تقليل التكرار البروتيني.
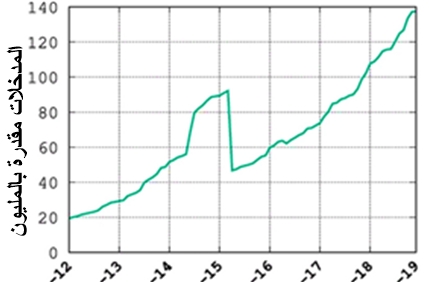
-انتشار الأخطاء الناتج عن ترابط قواعد البيانات، انتقال الأخطاء من قاعدة إلى أخرى.
-الخطأ في تحديد تتابعات النيوكليوتيدات، والتي تؤدي إلى صعوبة التعرف على الجينات وترجمة البروتين.
-تلوث تتابعات النيوكليوتيدات بسلسلة من ناقلات الاستنساخ المستخدمة لتضخيم قطع DNA.
-الأخطاء الأكثر شيوعاً في التتابعات تم إنتاجها قبل تسعينات القرن المنصرم، لذلك، يجب الانتباه إلى تواريخ تحديد التتابعات.
–ومن المشاكل الأخرى هي أخطاء الباحثين بسبب عدم التناسق والخلافات العلمية.
اتخذت بعض الخطوات لتوحيد تسمية الجينات عالمياً وكذلك إيجاد مفردات لُغوية موحّدة لوصف الجينات والبروتينات وهو ما أدّى إلى نشوء Gene ontology، ولمنع التكرار أنشأ المركز (NCBI) قاعدة بيانات مُشذّبة non-redundant (nr) خالية من التكرار تسمى (RefSeq)، تم فيها دمج التتابعات المتماثلة من نفس الكائن في إدخال واحد [6].
3-تحليل البيانات الحيوية وأهم خوارزميات المعلوماتية الحيوية
تأتي المعلوماتية الحيوية كمسار بحثي مساند لطرق البحث العلمية التجريبية لاختبار الفرضيات الإحصائية تطبيقياً. تعتمد جودة تنبّؤات المعلوماتية الحيوية على جودة قواعد البيانات وتعقيد الخوارزميات المستخدمة وكفاءة تلك الخوارزميات. تستخدم الخوارزميات كأداة هامة جداً في المقارنة بين التتابعات البيولوجية مثل تتابعاتDNA، وتتابعاتRNA، وتتابعات بروتينية. وتتم هذه المقارنة بغرض إيجاد التشابهات أو الاختلافات بين التتابعات المختلفة، واكتشاف الأصل المشترك لمجموعة جينات أو بروتينات، والتنبّؤ بالوظيفة والتركيب للجين أو البروتين المدروس. حيث يتم تقسيم مستويات التشابه إلى ثلاثة مستويات إحصائية كما في الشكل (3)، وهي مرتبطة بطول التتابع. علاقة مشكوك فيها (Twilight zone)، وتتابعات غير مرتبطة ولا علاقة بينها (Midnight zone)، أو علاقة معتبرة (Safe area) تتحدد أهميتها بحسب نسبة التشابه [2,7,8,9].
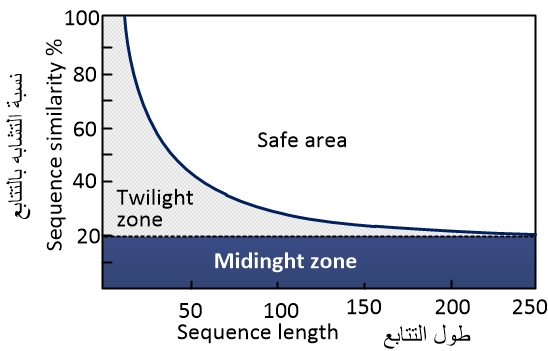
3- 1- خوارزميات المعلوماتية الحيوية
نعرض فيما يلي أهم الخوارزميات المستخدمة في المعلوماتية الحيوية:
أولاً: خوارزميات إيجاد أنماط التتابعات
يعد تحديد الأنماط Patterns ضمن التتابعات البيولوجية هو أكثر ما يحتاجه علماء المعلوماتية الحيوية، لأن هذه الأنماط في كثير من الحالات ترتبط بوظائف محددة للجزيئات الممثلة بهذه التتابعات، سواء كانت تتابع حمض نووي أو تتابع بروتين. إن وجود العديد من التكرارات في الكائنات الحية المعقدة (مثل البشر) يجعل لإيجاد النمط أهمية بالغة، من الخوارزميات المستخدمة في إيجاد النماذج الثابتة في التتابعات الضخمة [1] [10]:
– خوارزمية naïve لإيجاد الأنماط الثابتة: وتعتمد البحث الشامل، حيث تأخذ بالحسبان جميع احتمالات التتابعات الجزئية.
– خوارزمية بويير موور Boyer-Moore الموجهة: تستخدم بنية وهيكل النمط لتسريع البحث. وتعتمد على قاعدتين، قاعدة المحرف غير المطابق (bad-character rule)، وقاعدة اللاحقة الجيدة (good suffix rule).
– ماكنة الحالة المحددة (DFSM)، ونماذج ماركوف المخفية (HMMs).
ثانياً: خوارزميات الاصطفاف
خوارزميات الاصطفاف الشامل والموضعي، وتتشابه في الأساس وتختلف في استراتيجية تحديد الأمثلية في اصطفاف الأحماض الأمينية المتماثلة، وكلا النوعين يمكن أن يعتمد على واحدة من الطرق الآتية: طريقة مصفوفة التنقيط Dot matrix method، وطريقة البرمجة الديناميكية Dynamic programming method، وطريقة الكلمة Word method.
*مصفوفة التنقيط Dot matrix method:
وتعاني هذه الطريقة من التشويش والالتباس أو ما يسمى High noise level وخاصة عند مقارنة تتابعات طويلة [2,4].
*البرمجة الديناميكية Dynamic programming method
وترتبط بإمكانية تقسيم المشكلة إلى أجزاء مترابطة أبسط. من أهم الخوارزميات التي تعتمد البرمجة الديناميكية [11,12]:
- خوارزمية Needleman-Wunsch للاصطفاف الشامل Global alignment
- خوارزمية Smith-Waterman للاصطفاف الموضعي Local Alignment
- خوارزمية الاصطفاف شبه الشامل Semi global alignment
تعتمد الطرق السابقة على مصفوفات الاستبدال، التي تعطي الاحتمال الأقوى لاستبدال حمض أميني بآخر. يوجد نوعان أساسيان من مصفوفات استبدال الأحماض الأمينية، نوع يعتمد على خواص الحموض الأمينية، ونوع أشتق من الدراسات التجريبية. أظهرت الطريقة المعتمدة على خصائص الحموض الأمينية دقةً أقلّ مقارنة بالطريقة الثانية (الدراسات التجريبية). من المصفوفات المعتمدة:
- مصفوفة PAM) Point Accepted Mutation) وأهمها PAM250ـ[13].
- مصفوفة Block Substitution Matrices (BLOSUM) وأهمها BLOSUM62ـ[14].
*طريقة الكلمة Word method، وتتمثل بشكل أساسي بالبحث عن التشابه في قواعد البيانات
بسبب بطء البرمجة الديناميكية، وعدم فاعليتها في الكثير من الحالات، كانت هناك حاجة إلى طرق سريعة لمقارنة التتابعات. وأكثر الطرق شيوعاً هو استعمال الكلمة Word في حالة برنامج BLAST ويقابلها K-tuple في حالة برنامج FASTA.
-البرنامج BLAST : ومهمته إيجاد أعلى درجات التطابق للقطع بدون فجوات بين التتابعات المتقاربة، ويقدم دلالة إحصائية تساعد في التمييز بين التتابعات ذات العلاقة التطورية والأخرى المتباعدة أو الحاصلة بسبب الصدفة العشوائية [15].
-البرنامج FASTA: وهو أول برنامج استعمل للبحث عن التشابه بين تتابعات استعلام وقاعدة بيانات. بالإضافة إلى الدلالة الإحصائية في BLAST تستخدم في هذا البرنامج قيمة معيارية تربط تتابع الاستعلام مع قواعد بيانات أخرى بعيدة [16].
ثالثاً: اصطفاف التتابعات المتعددة MSA) Multiple Sequence Alignment)
لدراسة تتابعات ذات أصول مشتركة أي بينها علاقة تطورية ما، يتم اللجوء إلى اصطفاف عدد من التتابعات (MSA) وتعني صف ثلاث أو أكثر من التتابعات الحيوية. تستعمل درجات هذه الاصطفافات لبناء شجرة ثنائية تعكس العلاقة التطورية بين التتابعات، وتسمى بشجرة الدليل Guide tree. ومن الخوارزميات والطرق المستعملة للاصطفافات المتعددة [17,18,1]:
-طريقة الاصطفاف المتدرج Progressive alignment method
-طريقة الاصطفاف التكراري Iterative alignment methods
-طريقة القطع والتسلسل Divided and conquer alignment
-اصطفاف التراكيب Structural alignment
3- 2- العلاقات التطورية Phylogenetic relationships
العلاقة بين تطور الجينات وتطور الأنواع لا تكون متوازية، وذلك لأن تطور الأنواع وما يظهر عليها من أنماط يكون حصيلة تطورات وتغييرات لعدد من الجينات وليس جيناً واحداً. تستعمل مصفوفات المسافات [19] في عدد من طرق بناء الأشجار التطورية، وتأتي مصفوفات المسافات من مصادر مختلفة، مثل الدراسات المناعية، ودراسات المظهر الخارجي، وطول القطع الناتجة من تأثير أنزيمات القطع وغيرها من الأنزيمات، والمسافات الوراثية، وتمثّل الأخيرة عدد الطفرات أو التغيرات التطورية بين الأنواع منذ انفراجها عن بعضها. أبسط طريقة لحساب المسافة الوراثية هو حساب عدد الاختلافات بين اثنين من التتابعات، ولكن في بعض الأحيان تكون هذه الطريقة غير مجدية لعكس التاريخ التطوري للتتابعات كما هو موضّح في الشكل (4).
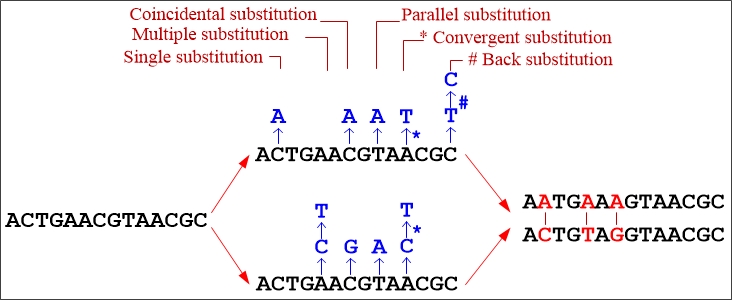
ومن النماذج الإحصائية التي تدرس هذه المشكلة: نموذج Jukes-Cantor model [20] ونموذج Kimura [21]. أما طرق وبرامج بناء الأشجار التطورية فهي متعددة [22,23,24,25]:
- طرق تعتمد على المسافات وتستخدم خوارزميات العنقدة Clustering algorithms.
- طريقة المعدل الرياضي غير المرجح(UPGMA)، وتعتمد على العنقدة المتعددة.
- طريقة ضم الجوار NJ) Neighbour joining).
- طرق الأمثلة Optimity-based methods، ومنها:
- (FM) Fitch-Margoliash
- التطور الأقل ME) Minimum evolution)
- الطرق المعتمدة على أساس الرمز Character-based methods
- طريقة الاقتصاد الأعظم MP) Maximum parsimony)
مشكلة العلاقات التطويرية الكاذبة:
ظهور فرع طويل في شجرة التطور، هو إحدى المشاكل التي ترافق بعض الطرق المستعملة، إذ يمكن أن تشير إلى علاقة تطويرية كاذبة، وذلك يعود إلى أن بعض الطرق المستعملة تفترض أن مسارات التطور متسايرة (تتطور بمعدلات متساوية)، وتأثير الطفرات متساوٍ في تحديد طول الأفرع، أو أنها تكون نتيجة لحدوث عمليات استبدال متكررة في مواقع محددة. من الطرق المستخدمة في تحديد الأشجار، طريقة بناء شجرة الاقتصاد الأعظم، وطريقة الاحتمال الأعظم Maximum likelihood، وطريقة الأحجية الرباعية [26] Quartet puzzling وطريقة الخوارزمية الوراثية Genetic algorithm [27]، وطريقة Bayesian [28].
معظم طرق إنشاء الأشجار التطورية لا تعكس الحقيقة كاملة. إذ من الشائع في الطرق التي تعتمد على المسافات وجود طفرات راجعة تمحو دليل التغير، بالتالي تكون بعض التغيرات غير محسوسة. لذلك عند تفسير النتائج يجب الأخذ بالحسبان النموذج المستعمل والافتراض الذي اعتمدت عليه الطريقة. يوجد طرق لتقييم طرق بناء الأشجار، أهمها: إعادة النّمذجة وإعادة التحليل، أو استعمال اختبار Jackknife [29]، أو التقييم المعتمد على البيانات، أو استخدام بعض المؤشرات الإحصائية مثل: اختبار الانحياز Skewness test [30]، واختبار التبديل Permutation test [31]، واختبار Bootstrapping [32].
4-الآفاق المستقبلية للمعلوماتية الحيوية
تعكس مجالات المعلوماتية الحيوية التي يجري البحث فيها وتطويرها حالياً حقيقة وجود تخصصات جديدة تدمج عدة مجالات، مثل الهندسة الطبية الحيوية والحاسوب والرياضيات والذكاء الصنعي والمعلوماتية الحيوية. ومن أهم هذه المجالات [33,34]:
-نمذجة بنية البروتين وتحليل وظائفه، وتمثل البروتينات الحسابية Computational proteomic.
-الجيل الجديد من التتابعات البيولوجية وتحليلها next generation sequencing
-الحوسبة الحيوية العالية الأداء أو التحليل الحيوي التّفرعي High performance in Bioinformatics.
-الطب الحيوي Biomedicine، الحوسبة الطبية الحيوية، والطب الشخصي.
-الهندسة الطبية الحيوية Biomedical Engineering، الجراحة بمساعدة الحاسب، والهندسة العلاجية
-نمذجة العمليات البيولوجية modelling biological processes.
-الرعاية الصحية والأمراض Healthcare and diseases، والصحة الالكترونية E-Health
-تطوير أدوات وبرمجيات جديدة Bioinformatics Tools and Software لتقديم وتحليل الجيل الجديد من التتابعات.
-المعلوماتية الحيوية في دراسة تطور علم الأحياء التطوري.
-تطوير شبكة الإنترنت على أساس المعلوماتية الحيوية ودعمها بأدوات مساعدة في فهم الأهمية البيولوجية للتتابعات الجينية في الأمراض.
-المعلوماتية الحيوية في المناعة (المعلوماتية المناعية) وتصميم اللقاحات.
-تصميم الأدوية بمساعدة الحاسب (Computer-Aided Drug Designing).
-المعلوماتية الحيوية في تحليل بيولوجيا الأمراض المستعصية مثل السرطان.
-المعلوماتية الحيوية في تحليل المجتمعات الميكروبية المعقدة الموجودة في مواقع الجسم المختلفة.
-نمو تقنيات الذكاء الصنعي AI وارتباطها بالمعلوماتية الحيوية بشكل كبير، حيث يتم استخدام الشبكات العصبونية وتقنيات التعلم العميق DL في تحليل التتابعات الجينية المعقدة.
-المعلوماتية الحيوية في معالجة الصورة Bioinformatics for Image Processing
-تطوير خوارزميات تحليل الكم الهائل من المعطيات Big Data في المعلوماتية الحيوية.
= = = = =
*الدكتور المهندس محمد ملحم – أستاذ مساعد في كلية تكنولوجيا المعلومات والاتصالات – جامعة طرطوس.
References
[1]. M. Rocha, P. G. Ferreira; Bioinformatics Algorithms/Design and Implementation in Python; Elsevier 2018.
[2]. S. Choudhuri; Bioinformatics for Beginners/ Genes, Genomes, Molecular Evolution, Databases and Analytical Tools; Elsevier 2014.
[3]. H. Patel, D. Bhatt, S. Shakhreliya, N. L. Lam, R.Maurya, V. Singh; an Introduction and Applications of Bioinformatics; Springer 2021.
[4]. R. Jiang, X. Zhang, M. Q. Zhang; Basics of Bioinformatics/ Lecture Notes of the Graduate Summer School on Bioinformatics of China; Springer 2013.
[5]. M. R. Bouadjenek, J. Zobel, K. Verspoor; Automated assessment of biological database assertions using the scientific literature; BMC Bioinformatics 2019.
[6]. K. Pruitt, T. Tatusova, D. Maglott; NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins; Nucleic acids research, 2005
[7]. J. XIONG; Essential Bioinformatics; Cambridge University Press 2006.
[8]. J.M. Claverie, C. Notredame; Bioinformatics for Dummies, 2ed edition, Wiley 2007.
[9]. B. Rost; Twilight zone of protein sequence alignments; Protein Engineering, Design and Selection, 1999.
[10]. J.Tarhio, E. Ukkonen; approximate boyer–moore string matching; SIAM Journal on Computing, 1993.
[11]. S. Needleman, C. Wunsch; A general method applicable to the search for similarity in the amino acid sequence of two proteins.J Mol Biol. 1970.
[12]. T. Smith, M. Waterman. Identification of common molecular subsequences.J Mol Biol 1980.
[13]. M. Dayhoff, R. Schwartz, B. Orcutt; a model of evolutionary change in proteins. Atlas of Protein Sequence and Structure, 1978.
[14]. S. Henikoff, J. Henikoff.; Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA1992.
[15]. S. Altschul, W. Gish, W. Miller, E. Myers. Basic local alignment search tool. J Mol Biol 1990.
[16]. W. Pearson, D. Lipman; Improved tools for biological sequence comparison.Proc Natl Acad Sci USA1988.
[17]. H. Carillo, D. Lipman; The multiple sequence alignment problem in biology. SIAM J Appl Math1988.
[18]. I. Wallace, O. O’sullivan, D. Higgins; M-Coffee: combining multiple sequence alignment methods with T-Coffee; Nucleic Acids Research, Volume 34, Issue 6, 1 March 2006,
[19]. W. Li; Simple method for constructing phylogenetic trees from distance matrices; Proceedings of the National Academy of Sciences, 1981.
[20]. T. Jukes, C. Cantor; Evolution of protein molecules; Mammalian protein metabolism. Academic Press, New York. 1969.
[21]. M. Kimura; A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences ournal of molecular evolution, 1980 – Springer
[22]. R. Xu, D. Wunsch; Survey of clustering algorithms – IEEE Transactions on neural networks, 2005.
[23]. P. Sneath, R. Sokal; Unweighted pair group method with arithmetic mean; Numerical Taxonomy, 1973.
[24]. N. Saitou, M. Nei; the neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 1987.
[25]. N. Saitou, T. Imanishi; Relative Efficiencies of the Fitch-Margoliash, Maximum- Parsimony, Maximum-Likelihood, Minimum-Evolution, and Neighbor-joining Methods of Phylogenetic Tree Construction in Obtaining the Correct Tree; The University of Chicago 1989.
[26]. K. Strimmer, A.V. Haeseler ; Quartet Puzzling: A Quartet Maximum-Likelihood Method for Reconstructing Tree Topologies; y the Society for Molecular Biology 1996.
[27]. D. Whitley; A genetic algorithm tutorial; Statistics and computing; Springe 1994.
[28]. J. Bernardo; Bayesian theory; AFM Smith 2009.
[29]. R. Miller; The jackknife-a review; Biometrika, 1974.
[30]. J. Bai, S. Ng ; Tests for skewness, kurtosis, and normality for time series data; Journal of Business & Economic Statistics, 2005.
[31]. D. Hillis, J. Bull; An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis; Systematic biology, 1993.
[32]. P. Mielke, K. Berry; Permutation methods: a distance function approach; Springer 2007.
[33]. O. Valenzuela, F. Rojas, I. Rojas, P. Glosekotte; Main findings and advances in bioinformatics and biomedical engineering IWBBIO 2018; BMC Bioinformatics 2020.
[34]. V. Singh, A. Kumar; Advances in Bioinformatics; Springer 2021.
جميع الآراء الواردة في هذه المقالة لا تعبّر بالضرورة عن رأي مركز جي إس إم وإنما تعبّر عن رأي صاحبها حصراً